Liabilities
This interactive dashboard combines a liabilities grid with a 3D structural viewer.
This tool is useful for identifying and addressing liabilities in antibodies. It is particularly useful for antibody engineering and developability optimization. AbLead utilizes an internal detection engine that incorporates the comprehensive Satlawa motifs (Satlawa et al., 2024) to identify sequence-based liabilities including deamidation, isomerization, and post-translational modifications.
The Liabilities method is a great example of my core work ethic to Be Inherently Lazy. Let the system find and map liabilities, display them on a structure, select them for mutation designs, and track the designs. Doing all of this manually takes hours while this tool completes the same tasks in seconds.
Accessing the Tool
Select at least one antibody in the Project View. Go to the Analysis menu and select Liabilities. This will open the Liabilities workspace in a new tab.
This analysis gives residue-level detail of the liabilities that were detected in the sequence(s) of the antibody. The liabilities are categorized by type and severity level. The severity level is determined by the potential impact of the modification on the antibody's stability, aggregation, or PTMs. Evaluation of the severity and structure position of the liabilities is critical for determining the best strategy for engineering the antibody to improve its developability.
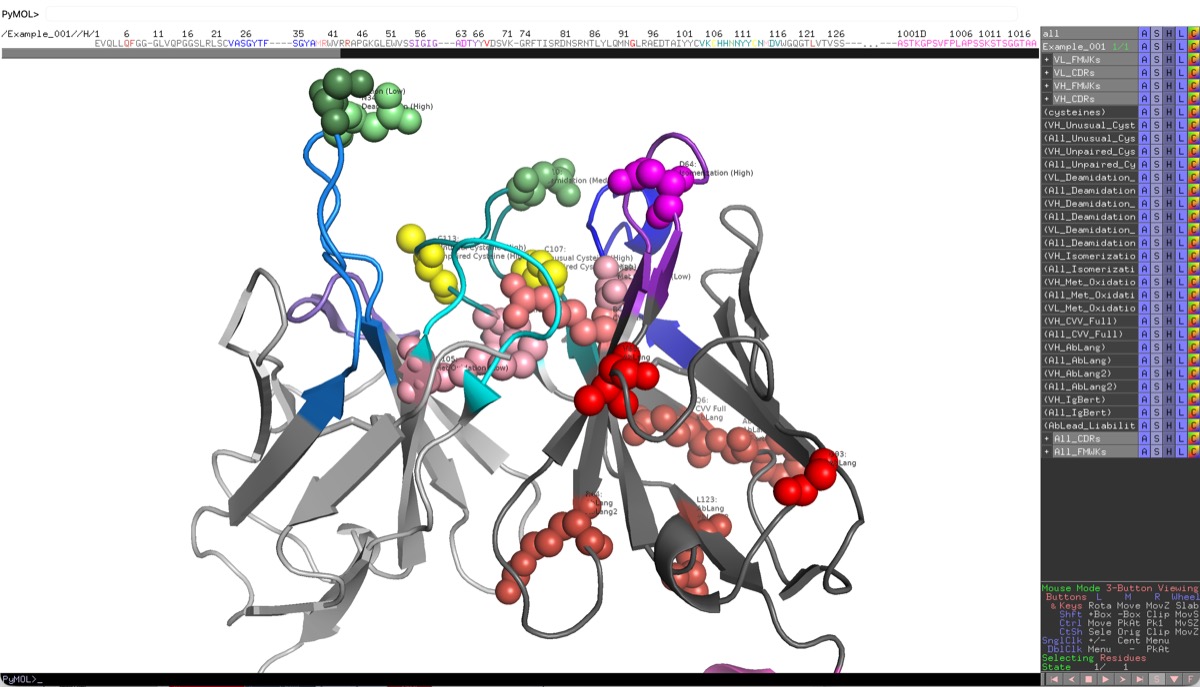
Select multiple entries to see them in the grid together. This allows for comparison of liabilities between different antibodies during evaluation and engineering. Selecting an entry name in the grid toggles the display of the 3D structure with liabilities highlighted. If the entry name background is light red, it means that no 3D structure is available for that entry. Toggle select liability rows to spacefill the associated residues on the 3D structure.
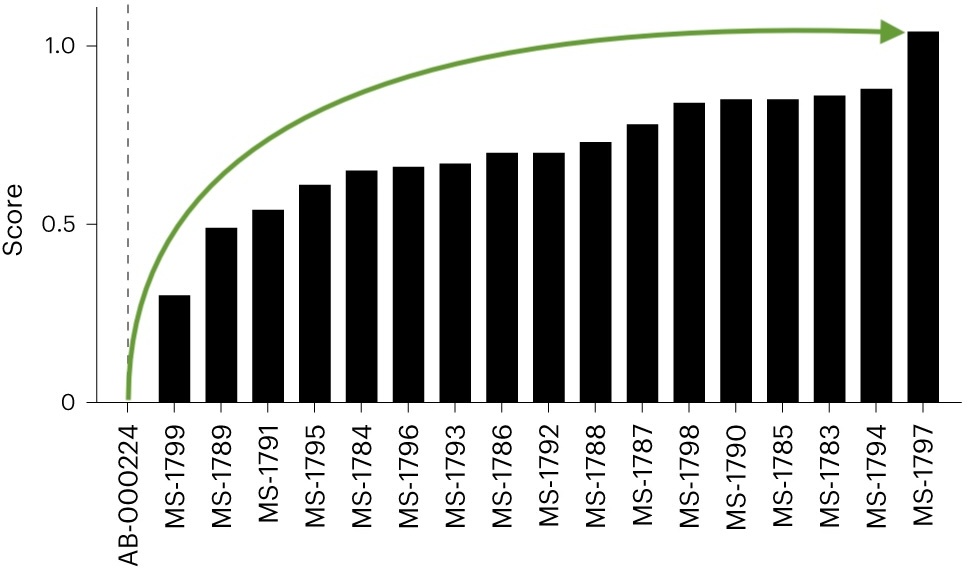
Engineering of strong stability violations has been shown to improve the developability of antibodies.
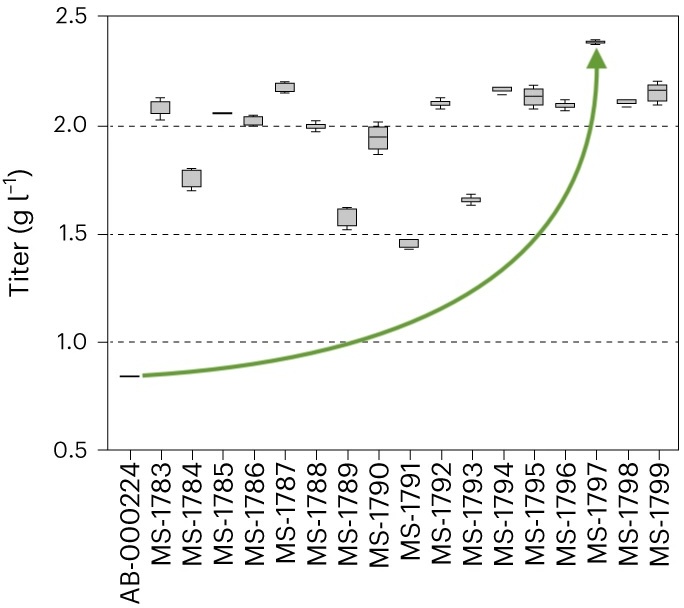
Stability engineering greatly improved overall biophysical properties...
... and more than doubled stable cell titer.
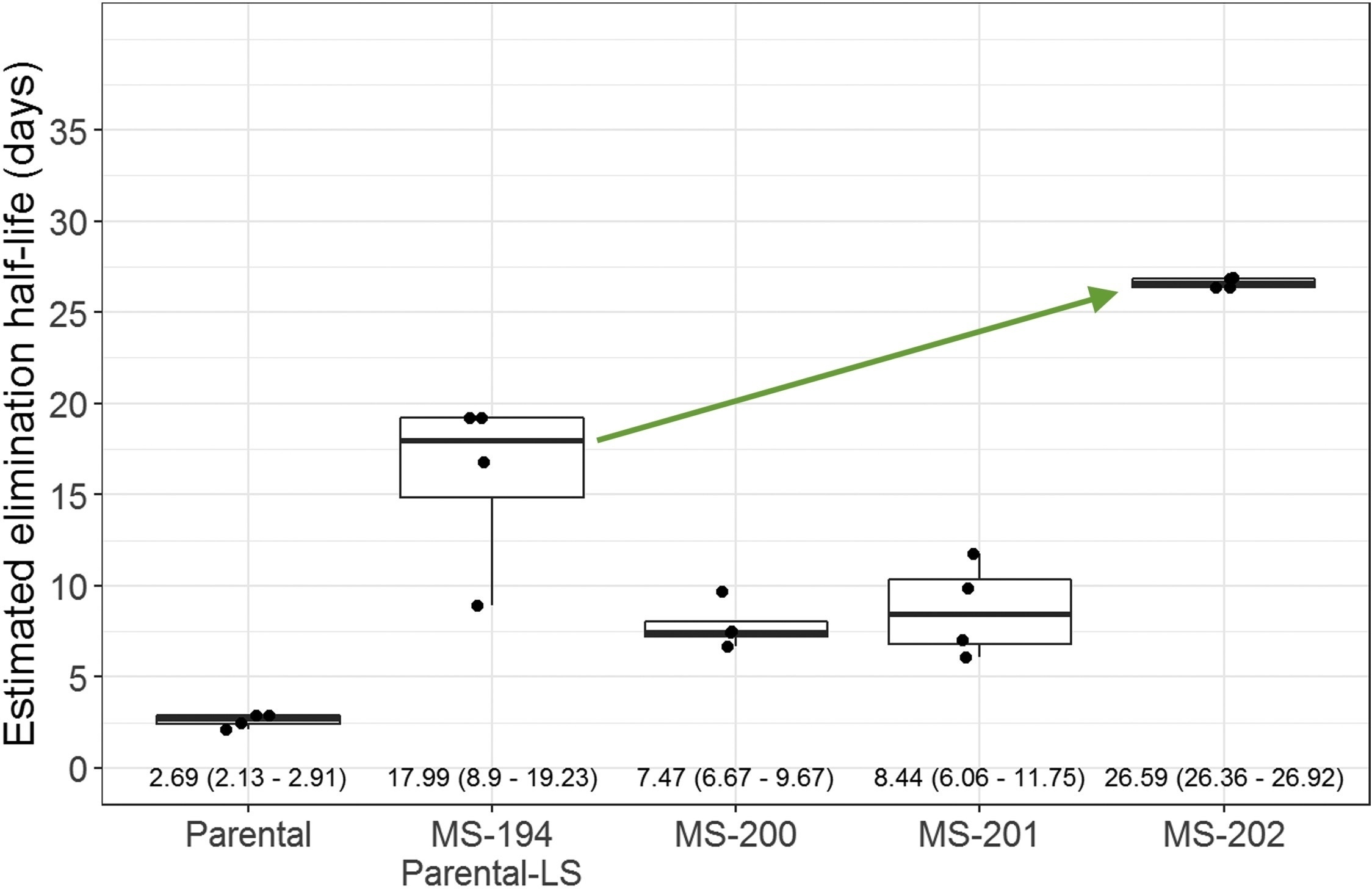
Developability optimization markedly improved serum half-life.
- Kerwin et al., "Framework Mutations of the 10-1074 bnAb Increase Conformational Stability, Manufacturability, and Stability While Preserving Full Neutralization Activity." Journal of Pharmaceutical Sciences (2020).
- Williams et al., "A candidate antibody drug for prevention of malaria." Nature Medicine (2024)
- Sajadi et al., "A comprehensive engineering strategy improves potency and manufacturability of a near pan-neutralizing antibody against HIV." Structure (2025)
Satlawa Liabilities Table
The following table summarizes the sequence-level liabilities detected by the AbLead internal engine, based on the Satlawa et al. (2024) classification.
| Name | Severity | Motif | Description |
|---|---|---|---|
| Deamidation (high) | High | N[GS] in CDRs | Deamidation of Asparagine occurs in the following motifs: NG motif (Asparagine followed by Glycine) and NS motif (Asparagine followed by Serine). Such motifs are known to be associated with deamidation (type of degradation) and can result in reduced "shelf-life". |
| Fragmentation (high) | High | DP in CDRs | Fragmentation occurs as cleavage at the interface between Aspartate and Proline. It is an example of a common motif that is susceptible to hydrolysis in response to pH. |
| Isomerization | High | D[DGHST] in CDRs | Isomerization of Aspartate occurs in the following motifs: DD (Aspartate followed by Aspartate), DG motif (Aspartate followed by Glycine), DH (Aspartate followed by Histidine), DS motif (Aspartate followed by Serine) and DT (Aspartate followed by Threonine). Such motifs are known to be connected to isomerization (type of degradation) and can cause a shorter "shelf-life" of antibodies. |
| Missing Cys (C) | High | C not present at 23 or 104 IMGT positions | Missing Cysteine occurs as cysteine absence at IMGT 23 or 104. Certain antibody sequence regions containing unpaired cysteines may result in structural changes, surface charges, or hydrophobicity. |
| Extra Cys (C) | High | C present at different position than 23 or 104 IMGT positions | Extra Cysteine occurs as cysteine present at a different position than IMGT 23 or 104. Certain antibody sequence regions containing unpaired cysteines can change an antibody's structure, apparent surface charges, or hydrophobicity. |
| N-linked glycosylation (NXS/T, X not P) | High | N[^P][ST] in variable fragment | N-linked glycosylation occurs as an addition of a sugar molecule. Reduced conformational stability and shorter "shelf-life" of antibody products are connected to asparagine linked glycosylation. Incidence of glycosylation in the CDRs can also directly impair antigen recognition and therefore lead to lower efficacy. |
| Deamidation (medium) | Medium | N[AHNT] in CDRs | Occurs in the following motifs: NA (Asparagine followed by Alanine), NH (Asparagine followed by Histidine), NN (Asparagine followed by Asparagine) and NT (Asparagine followed by Threonine). This type of deamidation is less common in comparison to the NG and NS motifs. |
| Hydrolysis | Medium | NP in CDRs | Hydrolysis gives rise to the DP motif as a result of the deamidation of Asparagine (N) to Aspartate (D). |
| Fragmentation (medium) | Medium | TS in CDRs | Occurs as pH-dependent cleavage at the Threonine-Serine interface. |
| Trp (W) oxidation | Medium | W in CDRs | Tryptophan oxidation is one of the Post-translational modifications (PTMs). |
| Met (M) oxidation | Low | M in CDRs | Methionine oxidation occurs in the CDRs. Reduced binding affinity and quicker degradation of the antibody product are linked to oxidation in these particular spots. |
| Deamidation (low) | Low | [STK]N in CDRs | Occurs in the following motifs: SN (Serine followed by Asparagine), TN (Threonine followed by Asparagine), and KN (Lysine followed by Asparagine). This type of deamidation is less common than others. |
Stability Cutoffs
AbLead incorporates three antibody-specific language models to identify anomalous residue positions (AbLang, AbLang2, and IgBert) as well as Covariance Analysis to detect co-evolutionary constraint violations. For language models, these calculate the residue likelihood difference (\(\Delta \ln(P)\)) between the model's preferred residue at a position and the actual residue in your sequence:
A higher difference indicates a sequence position that deviates from natural antibody distributions, making it a potential stability liability. Residues with likelihood differences greater than the Severe threshold are at a high risk of causing structural instability, low expression titers, aggregation, or poor developability.
For Covariance (CVV), violations represent pairs of residues whose amino acid biochemical classes do not match co-evolutionary constraints allowed in natural human germline V-genes.
Due to differences in calibration, their significance thresholds are scaled individually:
- AbLang (Original): Recommended cutoff is
3.0(Medium/Yellow) and5.0(Severe/Red). The original single-chain model has higher variance, meaning individual outliers often reach higher values. - AbLang2: Recommended cutoff is
1.0to1.5(Medium/Orange) and2.0(Severe/Red). As a paired model with germline bias correction, its probability predictions are tightly calibrated, making lower differences statistically significant. Differences above2.0indicate highly anomalous positions. - IgBert: Recommended cutoff is
1.5to2.0(Medium/Orange) and2.5(Severe/Red). This paired masked language model has intermediate variance; differences above2.5indicate highly anomalous positions. - Covariance Violations (CVV): Recommended cutoff is
3.0(Severe violation threshold). In residue-level views, individual violating positions are shown with their step score penalty, colored by severity: Low (1.0, Yellow), Medium (2.0, Orange), and Severe (3.0or higher, Red). Only overall CVV (CVV Full) is displayed at the residue level to align with the Covariance tool.
Overall Stability Score Thresholds
In addition to residue-level cutoffs, AbLead calculates overall stability scores for each antibody (Full Fv and Framework-only FR) by summing the likelihood differences. These scores are colored in the main results dashboard based on percentiles calibrated against the OAS reference database:
- AbLang Full: Green/Good \(\le 78.23\), Yellow/Warning \(\le 101.82\), Red/Severe \(> 101.82\)
- AbLang FR: Green/Good \(\le 39.45\), Yellow/Warning \(\le 59.55\), Red/Severe \(> 59.55\)
- AbLang2 Full: Green/Good \(\le 8.74\), Yellow/Warning \(\le 13.11\), Red/Severe \(> 13.11\)
- AbLang2 FR: Green/Good \(\le 4.58\), Yellow/Warning \(\le 8.03\), Red/Severe \(> 8.03\)
- IgBert Full (Paired Concatenation Mode): Green/Good \(\le 9.36\), Yellow/Warning \(\le 17.11\), Red/Severe \(> 17.11\)
- IgBert FR (Paired Concatenation Mode): Green/Good \(\le 4.33\), Yellow/Warning \(\le 10.32\), Red/Severe \(> 10.32\)
- CVV Full (Residue OMES): Green/Good \(\le 20.0\), Yellow/Warning \(\le 53.0\), Red/Severe \(> 53.0\)
- CVV FR (Residue OMES): Green/Good \(\le 1.0\), Yellow/Warning \(\le 7.0\), Red/Severe \(> 7.0\)
Single-Chain (VHH / Nanobody) Calculations
For single-chain sequences such as VHH or nanobodies, AbLang2 and IgBert overall stability and framework scores are reported as NC (Not Calculated):
- Paired Model Requirements: Both AbLang2 and IgBert are implemented in paired-chain mode within AbLead, which requires both the heavy and light variable domains to calculate joint probabilities.
- Missing Light Chain: Because VHH sequences lack a light chain variable domain, these paired models cannot execute, and their scores are marked as
NCrather than defaulting to0.00. - AbLang (Original): Unlike AbLang2 and IgBert, the original AbLang model executes using single-chain configurations and can calculate heavy-chain-only scores for VHH sequences.
Empirical Validation: Developability Thresholds (Jain Dataset)
Experimental characterization of the clinical Jain dataset (137 mAbs) shows that while linear correlations are moderate, there are pronounced threshold "drop-off" effects. Scoring above the 75th percentile of likelihood deviation for certain models indicates a dramatic loss of developability:
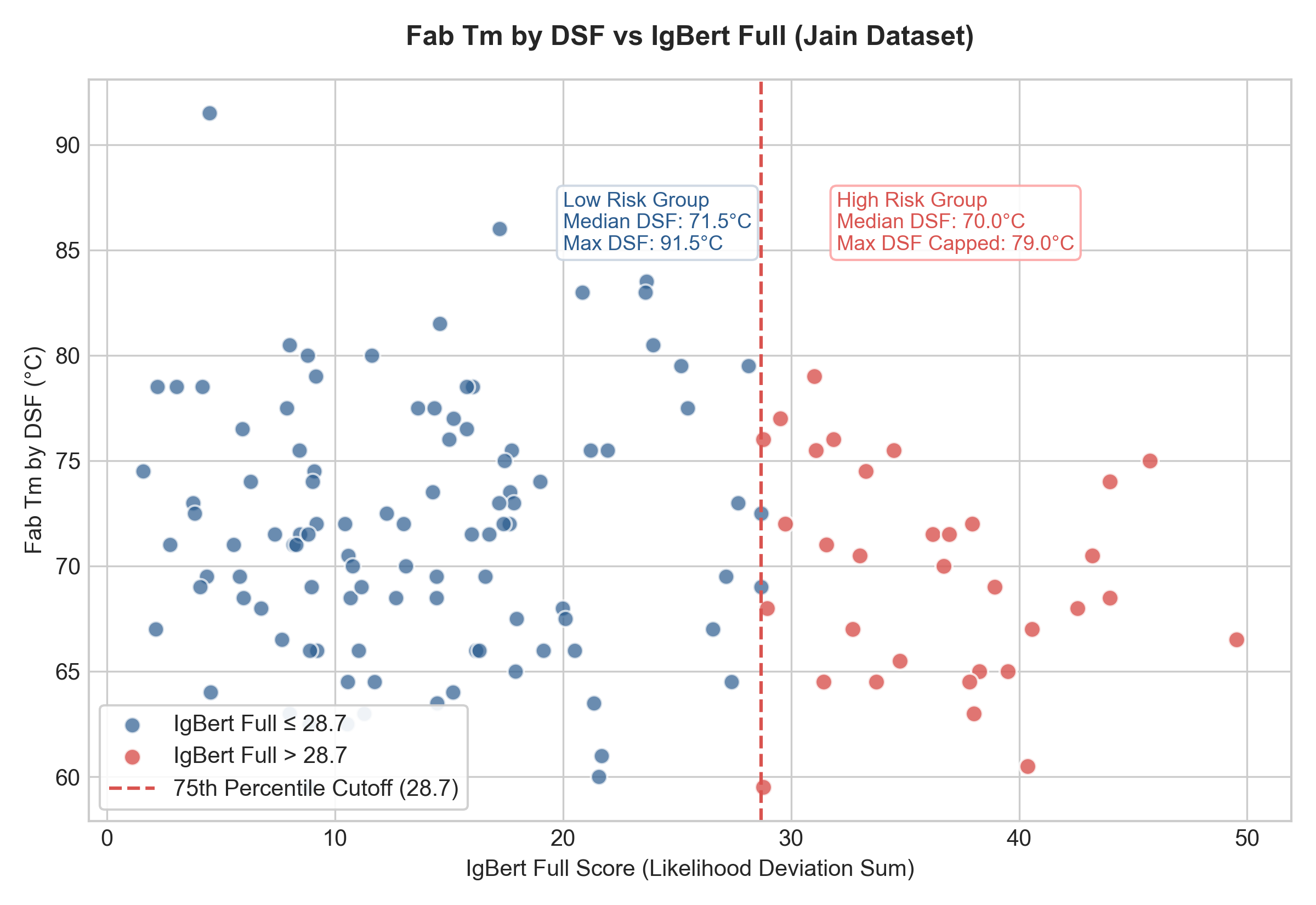
1. Thermal Stability (DSF) vs. IgBert Full
If the IgBert Full score exceeds 28.7 (75th percentile of the dataset), high thermal stability is essentially lost.
- IgBert Full \(\le 28.69\): Median DSF is 71.50 °C, with stable candidates reaching up to 91.50 °C.
- IgBert Full \(> 28.69\): Median DSF drops to 70.00 °C, and the maximum stability in this group is capped at only 79.00 °C (a complete drop-off of top-performing stable entries).
[!NOTE] This drop-off is not skewed by the extreme 91.5 °C outlier (tocilizumab, which has a highly natural IgBert Full score of 4.5). If this outlier is excluded, the low-risk group median remains 71.50 °C, the mean is 71.62 °C (compared to 69.62 °C for the high-risk group), the maximum stability reaches 86.00 °C (well above the 79.00 °C high-risk cap), and the difference remains statistically significant (Mann-Whitney \(p \approx 0.05\)).
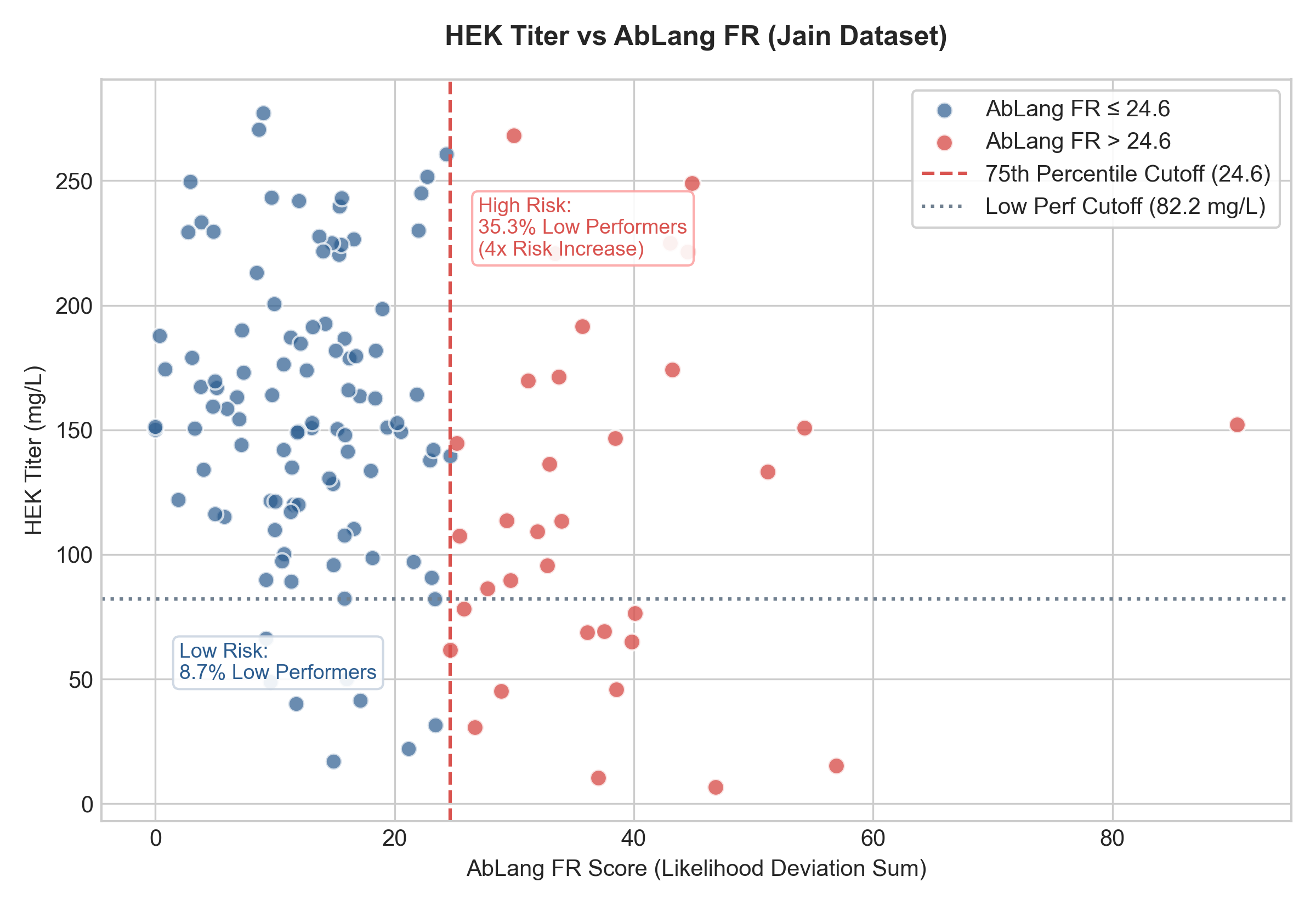
2. Expression Titer vs. AbLang FR
If the AbLang FR score exceeds 24.6 (75th percentile), expression levels drop significantly:
- AbLang FR \(\le 24.64\): Median Titer is 152.72 mg/L. Only 8.7% of these antibodies fall into the bottom 15% performance range (\(<82.2\) mg/L).
- AbLang FR \(> 24.64\): Median Titer drops to 111.32 mg/L, and the risk of poor expression increases four-fold, with 35.3% of these antibodies falling into the bottom 15% performance range.
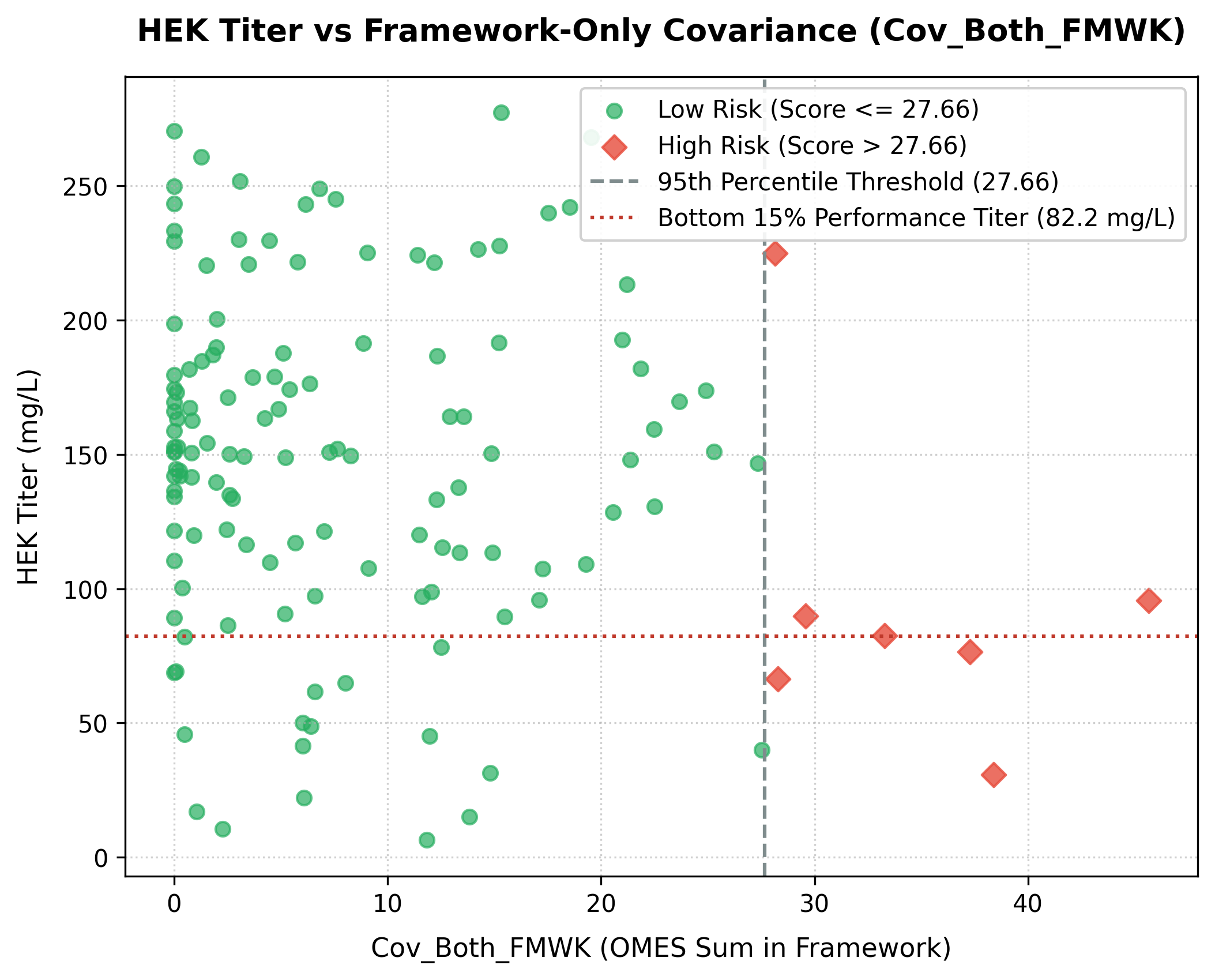
3. Framework-Only Covariance vs. Expression Titer
If the framework-only covariance score (Cov_Both_FMWK—summed only for violating position pairs where both positions reside in variable framework regions) exceeds 27.66 (the 95th percentile threshold of the dataset), expression titer drops significantly:
- Low Risk (
Cov_Both_FMWK\(\le 27.66\)): Median HEK Titer is 150.87 mg/L (mean: 149.96 mg/L). Only 13.8% of these antibodies fall into the low-expression category (\(<82.20\) mg/L). - High Risk (
Cov_Both_FMWK\(> 27.66\)): Median HEK Titer drops to 82.42 mg/L (mean: 95.19 mg/L), and the rate of poor expression increases three-fold, with 42.9% (3 out of 7) falling in the low-expression category. - Statistical Significance: The difference in titers between the low-risk and high-risk groups is highly significant (Mann-Whitney U test \(p = 0.0066\)).
Using the Tool
- Settings
- Select Severe: Selects liability rows which are considered severe.
- Hide Blank Entries: Hide entry rows which have no liabilities either before or after filtering.
- Chain: Select the chain to display.
- Region: Select the region to display.
- Liabilities: Select the liabilities to display.
- Stability Cutoffs: Set separate cutoffs for AbLang, AbLang2, IgBert, CVV Full, and CVV FR to control which residue-level stability and covariance anomalies are displayed.
Note: The liabilities in the grid after applying filtering are reflected in the downloads and structure view.
- Data Grid: Lists every detected liability (e.g., Deamidation sites, Unpaired Cysteines).
- Start/End positions.
- Severity Level (High/Med/Low).
- Liability Type.
- Positions that belong to the classic 30 Vernier zone residues (which may affect CDR conformation and antigen binding) are indicated with a (V) in the Region column.
- Engineering Mutation Designs: Click a liability to enter a mutation design used by the Engineering tab.
- Observations: Click a liability to enter an observation for that IMGT position. These are displayed in the Observations tab and on a clicked residue.
- 3D Viewer: If a PDB is associated, clicking Show 3D View will display the structure with frameworks, CDRs, and liabilities highlighted.
- Export
- Export Grid: Exports the liabilities to an Excel file.
- Export SVL: Exports the liabilities to SVL files, one for each entry, for use in the Molecular Operating Environment (MOE).
- Export PyMOL: Exports the liabilities to PyMOL scrips, one for each entry, for use in PyMOL.
- Indicator Dots: Dots on a residue position indicate that there is an associated engineering design or observation at that position. Engineering mutations will appear as a small eggshell square while observations will appear as a small orange circle.
References
- The PDB structure viewer is provided by Mol*.
- Satlawa et al., "LAP: Liability Antibody Profiler by sequence & structural mapping of natural and therapeutic antibodies." PLOS Computational Biology (2024).
- Hie, Brian L., Varun R. Shanker, Duo Xu, et al. “Efficient Evolution of Human Antibodies from General Protein Language Models.” Nature Biotechnology 42, no. 2 (2024): 275–83. https://doi.org/10.1038/s41587-023-01763-2.